At work yesterday, during lunch, I started a lengthy discussion that stretched out long beyond the boundaries of regular lunch hours. The discussion topic was my surprise that IT practices—particularly programming methodologies—hadn’t changed much, if at all, between my leaving the industry in 2007 and re-entering the industry in 2015. My surprise wasn’t a rethorical technique; my unpreparedness for this lack of change has lost me a long-running bet with Wiebe around 2007, when we stopped being colleagues for about 8 years. The content of that bet was as follows: I predicted that in 10 years time—now—computer science would have sufficiently evolved so that software would behave intelligently. In fact, I didn’t believe that AI would have emerged by now, but I did expect software architecture to have changed in such a fundamental way that computers would at least have behaved intelligent, even if they weren’t self-taught and self-learning.
I was doubly wrong when I entered the bet with Wiebe: software systems are messier and more difficult to change than they were 10 years ago, and special purpose, self-learning AIs are obsoleting humans even in such areas as driving cars. So, was I fundamentally wrong in thinking that a different type of software would be possible? Should I simply wait for AI to take over my job as a programmer and obsolete these concerns?
I don’t know. I do know that if AI doesn’t make my job obsolete (any time soon), I no longer believe that anybody else is going to make my job obsolete. And I really do believe that my job—the functions that I perform during the majority of my working hours—ought to be obsolete. But, since I can’t seem to convince anybody of this fact, I should suck up my pride and shame and start sharing what I’ve come up so far.
The notes that I’ve made the last few years aren’t as disjointed and messy as they were when I wrote my previous—and first—post about BULL. Here are some of the better-looking notes, from my A4 notebook:

BULL notes – A4 notebook p. 18

BULL notes – A4 notebook p. 19

BULL notes – A4 notebook p. 20

BULL notes – A4 notebook p. 21

BULL notes – A4 notebook p. 22

BULL notes – A4 notebook p. 23

BULL notes – A4 notebook p. 24
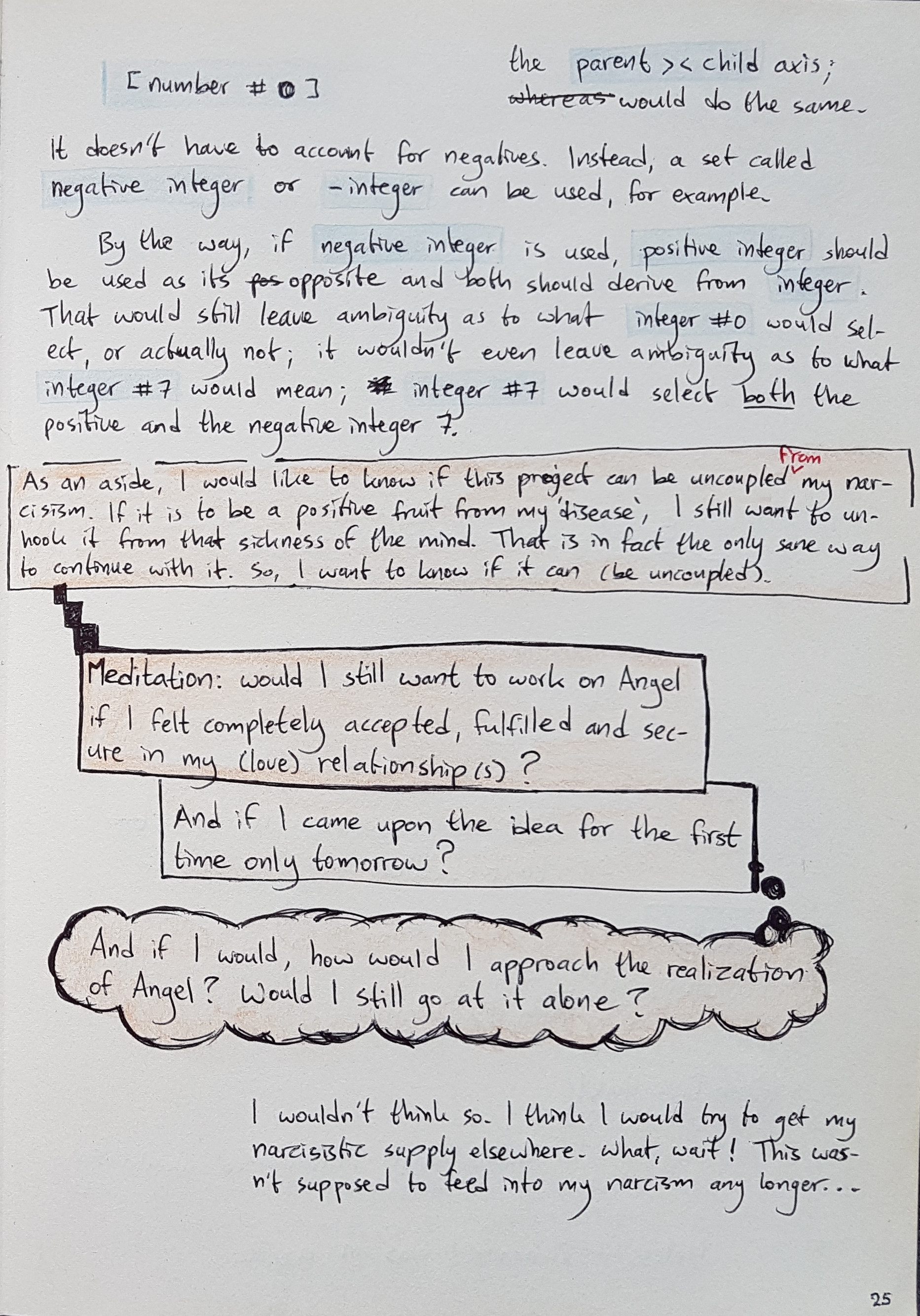
BULL notes – A4 notebook p. 25
Meanwhile, yesterday, while I should have been clocking work time, I’ve been condensing some of the threads in my head into a README.md in a newly published GitHub repository. This morning, I also rebased my latest half-assed attempt at a formal EBFN grammar and a parser in C++ to that same repository.

Recent Comments