Lectures
Energy metabolism
- “Explain the Harris-Benedict principle.”
Mean energy requirements are ~2000–2400 kcal/day, slightly less for ♀♀ than for ♂♂. Basal Metabolic Rate (BMR) can be estimated using the Harris-Benedict equation:
BMR = 10 x weight (kg) + 6.25 x height (cm) – 5 x age (yrs) + 5 if ♂
BMR = 10 x weight (kg) + 6.25 x height (cm) – 5 x age (yrs) – 161 if ♀According to this equation my BMR = 10 x 75 + 6.25 x 182 – 5 x 33 + 5 =
750 + 1137.5 – 165 + 5 = 1728 kcal/day.Multiply BMR with a number between 1.2 and 1.9, depending on daily activity level.
- “Name the energy-content and storage of three energy-carriers.”
Macronutrient class E (kcal/g) Carbs 4 Proteins 4 Fats 9 Alcohol 7 - “Outline the interaction between energy supplying metabolic pathways.”
Blood [glucose] has to remain ≈ 5mM. Lower levels will trigger the release of glucagon
- ≤ 1 day of fasting:
- glucagon (pancreatic alpha cells) → glycogenolysis (in muscle and liver tissue).
glucose (liver) → systemic release (through glucose-6-phosphate) - >1 day of fasting:
- triglyceride store,
gluconeogenesis by muscle catabolization → ketons
- blood [glucose] < 5mM
- glucagon ↑ (pancreas)
- blood [glucose] > 5M
- “Describe the regulation of the glycolysis/gluconeogenesis switch.”
Muscle tissue, at rest: Glycolysis (hexokinase) inhibited by its product [Glucose 6-Phosphate] increasing; pyruvate kinase inhibited by increasing [ATP]/[AMP] ratio.
Glucose → F-6P → F-1,2-BP + F-2,6-BP
Fructose 2,6-biphosphate activates PFK, thus upregulating glycolysisLow [ATP]/[AMP]: PFK ↑. PFK catalyses F6-P → F1,6-BP + ATP
Liver: no negative feedback of [Glucose 6-Phosphate] on hexokinase [because no hexokinase].Muscle tissue, during exercise:
Switch from glycolysis to gluconeogenesis occurs fructose-2,6-biphosphate
- “Name the most important steps in the Krebs cycle.”
First, glycolysis occurs in the cytosol:
- Pyruvate → Acetyl-Coenzyme A (CoA) (1CO₂ + 2e¯)
Krebs cycle / citric acid cycle happens within the mitochondrial matrix:
- C₂ (Acetyl-CoA) + C₄ (oxaloacetate)→ C₆citrate (2CO₂ + 8e¯)
- C₆ → C₅ + NADH + CO₂
- C₅ → C₄ + NADH + CO₂
- C₄ → C₄ (oxaloacetate) + NADH + FADH₂ + ATP
- “Describe the different components of oxidative phosphorylation.”
- A physical link to the citric acid cycle;
- four complexes (I-IV); and
- three proton pumps.
Inputs: 8 e¯ (carried by NADH⁺ and FADH₂ from the citric acid cycle) + 2O₂
Outputs: ATP + 4H₂O - “List the locations of the different metabolic pathways in the cell.”
glycolysis cytosol Krebs cycle mitochondrial matrix oxidative phosphorylation mitochondria
from Acetyl-CoA → 2CO₂ + 3NADH + FADH₂ + ATP:
8e¯ used to form 3NADH + FADH₂;
2C + 4O used to form 2CO₂
Fat metabolism
- “Describe the chemical structure of saturated and unsaturated fatty acids, of phospholipids, of triglycerides and of cholesterol.”
- Saturated fatty acids
- No double bonds between C atoms; all carbons are ‘saturated’.
- Unsaturated fatty acids
- One or more double bonds between (some) C atoms.
- Phospholipids
- Hydrophobic tail and hydrophilic head.
- Triglycerides
- chemically inert, energy-dense
- Cholesterol
- Many apolar structures (only 1 OH group), making it very hydrophobic.
- “Summarize how fatty acids are lysed, absorbed by intestinal cells and transported through the body.”
Lumen: triglyceride + lipase → FFAs + monoacylglycerol
Mucosal cells: reassembly of triglyceride → incorporated in chylomicrons
Lymph system - “List the characteristics, structure, and physiological role of 5 different lipoprotein particles.”
Lipoprotein particle Diameter (nm) Composition Physiological role Protein TG C CE PL Chylomicrons 500 2% 85% 1% 3% 9% Transport of fat to tissues VLDL 43 10% 50% 7% 13% 20% Carry newly synthesized triglycerides from the liver to adipose tissue IDL 27 18% 26% 12% 22% 22% Not usually detectable in the blood during fasting LDL 26–27 25% 10% 8% 37% 20% Transport of fatty acids and cholesterol from the liver to other tissues HDL 9.5, 6.5 55% 4% 2% 15% 24% Collect fat molecules from cells/tissues, and take it back to the liver - “Explain how the body mobilizes fat during fasting and how this is regulated.”
Glucogen triggers
Adipose cells: triacylglycerol (=triglyceride) → FFAs + glycerol
Liver cells: FFAs and cholesterol built into VLDL particles; glycerol → pyruvate (glycolysis), glycerol → glucose (gluconeogenesis)
Other tissues: FFAs oxidized → Acetyl CoA → citric acid cycle (→ CO₂ + H₂O) - “List the advantages of energy storage in the form of triglycerides.”
Triglycerides are very energy-dense: 9 kcal/g; hydrophobic; chemically inert; no important functional role; hydrophobic: the energy-equivalent of 12kg of fat in glycogen would require 100kg of water.
- “Point out the relationship between unsaturated fats, trans-fats and cardiovascular disease.”
unsaturated fats: VLDL↓ & HDL↑ => P(CVD)↓
trans-fats: LDL↑ & HDL↓ => P(CVD)↑
too much saturated fat: P(CVD)↑, P((colonic) cancer)↑, P(type II diabetes)↑
insufficient unsaturated fat: P(type II diabetes)↑ - “Explain the biological role of cholesterol.”
Cholesterol is essential for synthesizing cell membranes and for metabolites, including:
- steroid hormones (androgens, estrogens, progestins, glucocorticoids, and mineralocorticoids); and
- bile acids (detergent function for lipid absorption and biliary secretion, signaling function).
- “Describe how cholesterol synthesis is regulated.”
The molecular regulation of cholesterol synthesis follows a general feedback regulation system, wherein, when a precursor is converted into a product by an enzyme, decreasing [product] upregulates the enzyme (through a sensor) and increasing [product] downregulates the enzyme (through another sensor). The cholesterol sensor is SCAP (SREBP cleavage activating protein), which, in the presence of sufficient cholesterol, binds to an ER protein called Insig. SCAP escorts SREBP (sterol regulatory element binding protein) from the ER to the Golgi apparatus in the absense of cholesterol.
- “Define the differences between LDL and HDL.”
LDL transports fatty acids and cholesterol from the liver to other tissues.
HDL collects fat molecules from misc. tissues for transport back to the liver.[Learn diagram of damage by LDL and ‘repair’ by HDL.]
- “Describe the synthesis and metabolism of HDL.”
HDL is synthesized in the hepatocytes of the liver and in the enterocytes of the intestine. In these cells (almost exclusively), the Apolipoprotein A1 (APOA-I) gene is expressed, which is a major protein component of HDL. Cholesterol efflux to APOA-I is mediated by the ubiquitous ABCA1 transporter.
[And what about metabolism?]
- “Reproduce the cholesterol absorption route.”
exogenous C → chylomicrons (intestine) → VLDL → LDL → perihpial tissues
[Study slides]
- “Describe the complete reverse cholesterol transport route and the characteristics of the TICE route.”
Reverse cholesterol transport
Cholesterol in macrophages in periphial tissues is transported as HDL to the liver, which excretes it as bile into the intestine.
Transintestinal cholesterol transport (TICE)
TICE is a direct route of HDL from the intestine to the periphial tissues. It is an active payway facilitating the direct excretion of cholesterol from the enterocyte, likely mediated by apoB-containing lipoproteins.
- “Indicate why HDL particles are healthy.”
HDL
- promotes cholesterol efflux,
- promotes reverse cholesterol transport,
- inhibits LDL oxidation,
- inhibits endothelial inflammation,
- promotes endothelial NO production,
- promotes prostacyclin availability, and
- inhibits platelet aggregation.
Carbohydrate metabolism
- “List the different types of carbohydrates.”
- Monosaccharides: glucose, fructose, galactose;
- disaccharides: saccarose, lactose, maltose, isomaltose;
- polysaccarides: α-glucosides (starch, glucogen), β-glucosides (cellulose).
- “Summarize the digestion and transport routes of carbohydrates.”
Only monosaccharides are absorbed in the small intestine. Poly- and disaccarides first have to be split:
Starch → glucose;
lactose → galactose + glucose;
saccharose → fructose.Fructose absorbed by GLUT5 transporter;
glucose/galactose by SGLT1. - “Describe why and how blood glucose values are regulated.”
Insulin stimulates anabolic pathways (storage, synthesis, etc.), so that proteins are synthesized.
Glucagon stimulates catabolic pathways (breakdown, energy production), so that proteins are broken down. - “Explain the difference between glucokinase and hexokinase.”
Glucokinase (in pancreas, liver and brain cells) has a much lower KM (5.5 mmol/l) than hexokinase (in most other cells) so that insulin secretion of pancreatic B-cells can be positively correlated to blood plasma [glucose].
- “Describe the glycogen metabolism.”
Glycogen → Glucose 1-phosphate → Glucose 6-phosphate
Fasting (low glucose): glucagen released by α-cells in the pancreas. Glucagen stimulates the conversion of glycogen to glucose, as does adrenaline.
Eating (high glucose): insulin produced by β-cells in the pancreas prompts muscle and liver cells to convert glucose to glycogen.
- “Describe what it means to be lactose intolerant.”
Lactose-intolerent people have no lactase production or produce inactive lactase.
Protein and amino acid metabolism
- “Describe how proteins are degraded into amino acids.”
Dietary protein degradation
In the mouth: denaturation starts;
in the stomach: HCl and pepsine further break and hydrolize the proteins into polypeptides;
in the small intestine: proteases (e.g. aminopeptidase) break the polypeptides further down, into amino acids and dipeptides.
Intracellular protein degradation
Ubiquitinated protein → proteasome → peptide fragments + ubiquitin;
proteolysis: peptide framments → amino acids - “Indicate the difference between essential and non-essential amino acids.”
Essential amino acids cannot be synthesized by the body and have to be included in dietery sources.
- “Indicate the location of protein degradation.”
In the mouth, the stomach and the small intestine. Also, intracellularly.
- “Describe the glucose-alanine cycle.”
In the muscle: glucose →(glycolysis)→ pyruvate; branched-chain amino acids → NH₄⁺; pyruvate + NH₄⁺ → alanine
Alanine is transported to the liver, where it can be used in gluconeogenesis (alanine → pyruvate → glucose) or converted to urea (alanine → glutamate → NH₄⁺ → urea). - “Describe the urea cycle.”
Amino acid degradation takes place mostly in the liver: alpha-amino acid → glutamate → ammonium (NH₄+) → Urea (with NH₂ groep). (Urea cycle takse two NH₄+ and Fumerate [Gluconeogenesis lecture]. Fumerate is another of the amino acid breakdown products (not in the urea cycle).)
- “Describe how amino acids are used as an energy source.”
Gluconeogenesis: CO₂ + NH₄⁺ → urea cycle. In one of the next steps, aspartate (one of the products of an alpha-amino acid + oxaloacetate) enters the cycle: citrulline + aspartate → arginino-succinate → fumarate + arginine.
fumarate → malate → oxaloacetate → glucose + aspartate. - “Describe situations in which there is an abnormal protein requirement.”
- Infancy
- Pregnancy
- Malabsorbtion of amino acids
- Metabolic disease (e.g. PKU/-phe)
- Protein sensitivity / bovine protein intolerance
- Diseaese/trauma/wounds
Metabolic regulation
- “List the transcription factors and associated ligands which are important for the regulation of the energy metabolism.”
LXR (liver X receptor) is activated by cholesterol. In the liver, LXR activates ABCG5/8, inceasing the release of Chol+BA by hepatocytes. This, in turn, lowers blood cholesterol, as well as atherosclerosis, but also raises triglyceride production. LXR is thus a promising target for treating atherosclerosis, with the unintended side-effect of weight-gain. Indeed, a synthetic LXR ligand (GW3965) has been found to inhibit the development of atherosclerosis in LDLR and apoE knockout mice by S.B. Joseph et al. (PNAS 2002;99:7604-7609). And, indeed, the treated mice gained weight as well.
LXR also stimulates the absorption of FFA by the hepatocytes.FXR (farnesoid X receptor) is activated by bile acids. FXR then activates MDR2 and BSEP transporters (which facilitate the release of PC and BA, respectively). FXR lowers cholestasis, the incidence of chol. gallstones, and the production of triglycerides; as a target for weight-loss, the unintended side-effect will be atherosclerosis.
PPAR (Peroxisome Proliferator-Activated Receptor) family of receptors bind to fatty acids. PPARγ is implicated in energy storage. PPARδ and PPARα are implicated in energy burning.
- “Explain how a nuclear hormone receptor works.”
Ligand-mediated activation: nuclear receptor (NR) binds to HRE (Hormone Receptor Element), a stretch of DNA to which also an RXR nuclear receptor is bound. This complex is bound by a HDAC co-repressor complex, actively repressing the HRE. When a ligand binds to the NR, the HDAC co-repressor complex is displaced by the HAT co-activator complex, which causes acetylization of a DNA region adjacent to the HRE. (HDAC = Histone Deacetylase; HAT = Histone Acetyl Transferase.) Acetylization activates the genes.
- “List the characteristics of the metabolic syndrome.”
Obesity, high blood sugar, high LDL, high blood TG, high blood pressure.
- “Name drugs that act through the activation of nuclear hormone receptors.”
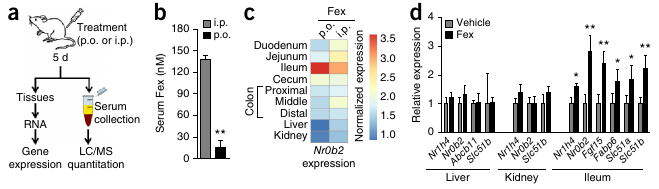
- Fex selectively activates intestinal FXR.
- Avandia activates PPARγ, causing the burning of FFAs.
- “List processes that take place during the enterohepatic cycle of bile salts.”
FXR is expressed in the liver, which releases bile salts. Bile, produced by hepatocytes (liver cells) ends up in bile ducts, aided by 3 ABC transporters of relevance: ABCG5/8 transporters (for cholesterol), MDR2 transporters (for PC=phosphatidylcholine), and BSEP transporters (for BA). Bile is made up of 4% cholesterol, 24% PC and 72% BA. PC helps to neutralize BA so that it doesn’t damage cell membranes.
BA reenters the cell with the aid of a transporter called NTCP. Cholesterol also enters the hepatocytes (liver cells) (LDL through LDLR, HDL through SR-B1).
FXR activation triggers the release of bile from the hepatocyte. In enterocytes (in the illeum, the final section of the small intestine), FXR downregulates the uptake of BA by ASBT transporters and stimulates the release of BA by OSTa/b. FXR is also responsible for the release of bile salts from the illeum into the capillaries. Bile salts are also released into the blood stream by the gallbladder, which constricts under the influence of CCK.
- “Decribe the regulation of fat metabolism in the liver.”
Triglyceride (TG) from the diet or adipose tissue is hydrolysed (lipolysis) into free fatty acids (FFA), which enters the hepatocyte through the CD36 transporter. FFA in the hepatocyte can be converted into TG and stored in a complex with cholesterol, or TG+chol. can leave the hepacotye through MTTP as VLDL. Alternatively, FFAs can be used in the FAO pathway for the energy necessary to produce ketone bodies during the production of Acetyl CoA.
LXR activates lipogenesis (FFA synthesis from sugars) and increases FFA uptake from the blood, causing more TG production, which is dependent on [FFA]. ABCG5/8 transporters are also activated, increasing the release of Cholestorol and BA into the bile ducts. FXR, however, inhibits LXR and also directly inhibits lipogenesis.
- “Describe the transcriptional regulation of different muscle fiber types.”
PPARδ function remained unknown for a long time. It didn’t help that knockout mice died while still in the embryoic phase. The solution was to construct mice (VP16-PPARδ mice) in which PPARδ is only locally overexpressed, in muscle. VP16-PPARδ red muscle mass increased relative to controls.
Transgenic mice with increased PPARδ in their muscles were true “marathon mice”. These mice are also overweight-resistent, which makes PPARδ a promesing anti-obesity drug-target. It as already used as doping by cyclists.
PPARδ expression in muscle tissue → fast twitch,
PPARδ suppression in muscle tissue → slow twitch. - [Study Slide 49 & 50.]
Integration of metabolism
- “List different satiation signals.”
- GLP-1, secreted by L cells in the small intestine, signals satiety in the brain; as does
- CCK, which is also released by the small intestine.
- Leptin is an adipokinese, a signal molecule from fat tissue.
GLP-1 also causes the release of insulin by the pancreas.
CCK also causes the constriction of the gall bladder. - “Indicate how leptin regulates the energy balance.”
Normally, a decrease in fat cell mass → decrease in leptin expression → decreasing leptin action in hypothalamus → increase in food intake, while an increase in fat cell mass → increase in leptin expression → increasing leptin action in hypothalamus → decrease in food intake.
- “Indicate the consequences of leptin resistence and deficiency.”
Leptin-defficient (ob-/ob-) mice and men never stop eating.
- “Indicate the consequences of excessive calorie intake.”
Metabolic syndrome occurs when an excess of triacylglycerols can no longer be stored by the adipose tissue and is stored as lipid drops in other tissues. Insuline resistance results from energy stress (overnutrition and inactivity). Stress-induced serine kinases (which increase through the effects of mitochondrial overload and the increase of DAG and Ceramide) make GLUT4 less easily activated by the insuline receptor in the plasma membrane.
Blood glucose rises with insulin resistence, which makes the pancreatic beta-cells work harder to produce more insuline, until, finally, the pancreatic cells give in and Type II diabetes turns into Type I diabetes. (During pregnancy, some measure insulin resistence is required. Maybe a contributing factor in insulin resistance is increased estrogen production by adipose tissues?)
- “Describe the changes in energy metabolism during fasting.”
The liver mobilizes the glycogen store: Glycogen → G1-P → G6-P → Glucose. (In the muscles and brain, glycolysis produces pyruvate from G6-P. Pyruvate is burned into CO₂ and H₂O in the brain and in aerobically exercised muscle, and into lactate in anaerobically exercised muscle.) However, this is only enough for 1 day.
Gluconeogenesis uses amino acids to produce new glucose. Burning too much protein for energy is dangerous, though. The second priority is thus to maintain proteins. This is to some extent solved by ketone bodies [Fig 27.12/13], which can be formed from free fatty acids. Ketone bodies are hydrophilic and can pass the blood-brain barrier. Red blood cells still require glucose which is why the blood [glucose] has to remain > 2.2 mM.
Tutorials
Common objectives for all articles:
- “List the most important conclusions from the articles treated.”
- “Understand the principles upon which the articles are based.”
Diet
- “Explain why specific metabolite concentrations have been measured in the study.”
Blood [creatine] as an indicator of renal health;
Blood [C-reactive protein (CRP)] as an inflammation marker;
Urine [ketone] as an indicator of a glucose shortage, thus the burning of fat. - “Describe which metabolic adjustments take place in reaction to a low-carb or a low-fat diet.”
In both studies,
down: body weight, fat mass, triglyceride, CRP, 10-year Framingham CHD Risk Score;
up: HDL,
but for the people in the low-fat group everything goes up/down further. - “Formulate a follow-up study to the diet study.”
Is het “laag-vet dieet” in deze studie wel een laag-vet diëet? Het zit nog steeds boven de aanbevelingen van het voedingscentrum.
Measure BMR to check if low-carb increases resting energy expenditure and total energy expenditure.
Cardiovascular disease
- “Describe where and how TMAO is produced.”
phosphatidylcholine (PC) / choline → TMA → TMAO
TMA (gasseous) is produced from PC/choline by intestinal microflora.
TMA is converted to TMAO by FMO3 enzyme. - “Describe the possible mechanism how phosphatidylcholine influences the formation of artherosclerosis.”
“Aortic macrophage content and scavenger receptor surface within aortic lesions were markedly increased in mice on the high-choline diet” (Wang et al. 2011).
- “Point out possibilities of dietary interventions to lower the formation of TMAO.”
Eat less red meat and other sources of choline.
Metabolic regulation
- “Explain the difference between brown and white adipose tissue and their functions.”
There is higher energy expenditure in brown adipose tissue (BAT), which is caused by the greater amount of mitochondria than in white adipose tissue (WAT).
- “Point out the role of UCP1 in adipose tissue.”
UCP1, which is expressed in BAT mitochondria, deflects the energy in the proton gradient to the production of warmth instead of ATP.
- “Describe the effect of beta-adrenergic receptor (βAR) activation on fat cells.”
The authors report increased UCP1 expression in adipose tissue. This might (partially) have been induced by increased beta adrenergic receptor (βAR) expression in WAT, through activation by catacholamine, which also explains the increase in FFAs and the decrease in triglycerides.
- “Explain what the respiratory exchange ratio (RER) tells about energy usage and how you can measure this.”
RER, which is the ratio VCO₂/VO₂, provides information on the active metabolic pathway. RER = 0.7 during the oxydation of carbohydrates and RER = 1.0 during the oxydation of fatty acids. RER ≤ 1.0. During the night, when most humans are asleep, we burn more fat than carbohydrates. For mice, which are nocturnal, the opposite is true.
These values would have been supplied by the apparatus with which they performed energy expenditure measurements: a Comprehensive Lab Animal Monitoring System (Columbus Instruments).
- “Explain what insulin and glucose tests are and what insight they provide into glucose homeostasis.”
Hyperinsulinemic euglycemic clamp study: Mice are infused with glucose and the insuline response is measured. […]
Food and epigenetics
- “List 3 basic epigenetic mechanisms.”
- DNA methylation,
- histone modification, and
- non-coding RNA.
- “Eplain how DNA methylation can affect gene expression.”
DNA methylation—the addition of a methyl group to cytosine or guanine nucleotides—affects the readability of the methylized DNA.
- “List human examples of nutrient-epigenetic interactions (historic or geographic).”
- Dutch Famine: children of mothers who suffered malnutrition during pregnancy showed an increase change of developing a number of metabolic diseases.
- Barker Study in Wales: lower birth weight increased the chance of mortality through heart disease.
- Gambian women: higher methylation of 6 metastable epialleles (MEs) in children conceived during (the meager) rainy season.
- “Discuss the experimental planning of a diet/epidemiological study.”
[…]
Nutrition and microbiota
- “List the effects of a vegetarian and an animal-based diet on the microbiome and its fermentation products.”
“The animal-based diet increased the abundance of bile-tolerant microorganisms (Alistipes, Bilophila and Bacteroides) and decreased the levels of Firmicutes that metabolize dietary plant polysaccha-
rides (Roseburia, Eubacterium rectale and Ruminococcus bromii). Microbial activity mirrored differences between herbivorous and carnivorous mammals2, reflecting trade-offs between carbohydrate and protein fermentation.” (David et al. 2014) - “Describe the relationship between the microbiome and obesitas.”
Obesity of mice is partly determined by gut microbiota; if these microbiota are transplanted, the recipient mice will also become obese (Ley et al. 2006): “the relative proportion of Bacteroidetes [relative to Firmicutes] is decreased in obese people by comparison with lean people, and […] this proportion increases with weight loss on two types of low-calorie diet.” David et al. (2014) observed a reduction in weight for subjects that switched to an animal-based diet.
More fermentation → more FA extracted from food.
- “Indicate how the microbiome contributes to the development of children in relation to disease and what the role of nutrition is in this process.”
[…]
Nutrition and microbiata learning objectives from slides
- “For four types of dietary fiber, list the effect on the microbiome, and on FA/butyrate production.”
- Inuline;
- resistant starch;
- cellulose;
- Gos/Fos (oligosaccharides).
- “[…]”
Eatmeter
- “Calculate the daily energy requirement.”
\(
BMR_♂(w=75, h=182, a=33) = C_♂ + W_♂w + H_♂h – A_♂a
\)Depending on physical activity level, multiply BMR with a value between 1.2 and 1.9.
- “List factors which influence the basal metabolic rate.”
Age; gender; physical activity level.
- “List the physiological function of dietary fibers.”
- stommach filling;
- bulk forming and nutrient dilution causing slower absorption in small intestine and less glucose spiking, while food uptake remains efficient;
- fiber-nutrient interaction;
- longer transit time;
- a food source for microbiota.
- “Indicate by which pathways the microbiome can influence the energy metabolism.”
[See my notes on the Eatmeter tutorial.]
- “List the fat-soluble vitamins and water-soluble vitamins, and explain the difference in terms of absorption, transport and storage of these vitamins.”
Fat-soluble: A, D, E, K.
Water-soluble: B, C.Absorption of fat-soluble vitamins goes up with increasing dietary fat uptake.
Caroric restriction
- “Define ‘ageing’ and interpret the term in an evolutionary context.”
Senescence: declining function with advancing age, among which decreasing fertility, and increasing mortality.
Ageing is not universal. Hydra, for example, do not age. Hydra reproduce asexually, which may hint at the absense of a disposable soma. Because Hydra genes do not invest in genetic variation, reproduction may confer no advantage over simply staying alive. - “Explain ultimate and proximate ageing theories, and give examples.”
Ultimate ageing theories explain why evolution has produced organisms that age, in terms of phylogeny and functional adaptation; while proximate theories explain how organisms age, in terms of ontogeny and causative mechanisms.
The distinction between proximate and ultimate ageing theories is meaningless. Most ageing theories attempt to explain both proximate and ultimate questions. Evolutionary ecologists have to be concerned with all 4 of Tinbergen’s questions.
A recurring concept in ultimate ageing theories is that wild animals do not often live to an age at which senescence comes into play, which severely limits the ability of natural selection to act on traits which would inhibit senescence.
- Mutation accumulation theory: senescence can persist in a population simply because natural selection doesn’t get a chance to come into play due
to the low likelihood of individuals surviving long enough to reap a fitness benefit from decreased
senescence. Then, old-age is caused by late-acting deleterious mutations which had no influence on reproduction (Medawar, 1952). - Antogonistic pleiotropy: some late-acting deleterious genes may offer a benefit early in life, thus being favored by selection, even if selection forces are still active in later life (Williams, 1957). The slides say that the theory considers “no selection pressure on detrimental effects in postreproductive lifespan,” but this is false; it is definitely a theory of trade-offs between alleles that give an advantage during reproductive life and alleles that give an advantage during late life.
- Disposable soma: life-history trade-off between growth/reproduction and somatic maintenance/repair (Kirkwood, 1977).
- Programmed death / phenoptosis / acute senescence: a possible example are Pacific Salmon, which spawn immediately after spawning. Although this is normally assigned to exhaustian, Sapolsky (2004) points out that if a Salmon’s adrenal glands are remove right after spawning, it will live for quite a bit longer. Similar observations have been made in some species of Australian marsupial mice.
- Mutation accumulation theory: senescence can persist in a population simply because natural selection doesn’t get a chance to come into play due
- “Define the term ‘calorie restriction’ (CR).”
CR is a 10–30% reduction in calories, below what would be eaten when feeding ad libitum.
- “Apply principles outlined in ageing theories to the topic of caloric restriction.”
- “Explain major differences between small lab animals and primates, which may lead to different responses to CR.”
Small rodents are r-strategists, many offspring, short weaning-times, little energy invested per individual, short life-spans.
Primates are K-strategists: few offspring, long weaning-time, lots of inergy invested per individual offspring, long life-spans.Compared to rodents, somatic maintenance and repair in primates is subject to much stronger and sustained selection forces, because parents have to grow old enough to take care of their young. This produces very different outcomes when considering disposable soma models. Additionally, grand-parental care can play a role, so that kin-selection must also be considered.
- “Explain major considerations in the setup of calorie restriction studies in primates, especially regarding the measurement and implementation of CR.”
[…]
- “Explain different setup and observations in the NIA and UW CR studies, and outline possible explanations.”
NIA UW Study onset 1987 Control group diet rationed ad libitum Supplementation control and CR CR only Age-related mortality no reduction sign. reduction Metablic health / overall function + Immune response improved in young-onset monkeys - “Explain the importance of control groups in CR studies.”
Ehm, where else are you going to compare the CR treated subjects with?
- “Name further factors, in addition to CR, that affect life and health span.”
Genetics. Wild-caught mice showed no increase in life-span due to CR. Also worth mentioning that the NIA population was more genetically diverse.


Recent Comments