Tutorial no. 6 for the RuG Metabolism & Nutrition course builds on the lecture [Lecture 5] on the same subject by Hans Jonker. It is also the subject which me and two others have to give a presentation on this Friday for the rest of Group C to initiate the group discussion. The article which is to be discussed during our presentation and the rest of the tutorial is a 2014 Nature publication [doi:10.1038/nm.3760] by Sungsoon Fang et al., titled “Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance.”
For this tutorial, the lecturer requested that we answer the following question about the article under discussion, which I might just as well, because it could be a good vantage point for preparing our editorial and presentation.
- “What is the scientific relevance of this article, summarized in a single sentence?”
In the profitable quest for “treatment of obesity and metabolic syndrome”, by using an agent that selectively targets intestinal bile acid sensor receptors (FXR), Fang et al. have demonstrated that they could reduce “diet-induced weight gain, body-wide inflammation and hepatic glucose production, while enhancing thermogenesis and browning of white adipose tissue.”
I say “profitable” because six of the paper’s authors may be entitled to royalties in the event that the FXR molecules and/or methods of use, which they co-invented, are commercially developed.
- “What are the specific finds? Mention at least 3.”
- “Orally active Fex shows minimal systemic exposure.”
Fexaramine (Fex), the FXR agonist used in the study, is poorly absorbed in the small intestine, and can, as such, be orally administered to selectively activate intestinal FXR. This sidesteps the negative side-effect (exacerbated weight gain and glucose intolerance) of FXR activation in the liver. [These effects were also mentioned in Lecture 5.]
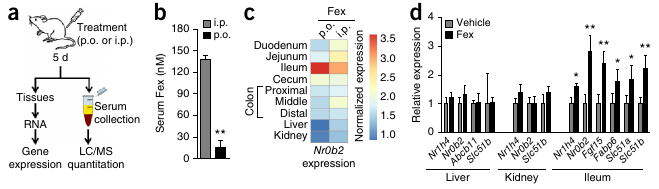
Figure 1 from Fang et al. 2014 shows that (b) Serum [Fex] stay low when Fex is administered through the oral gavaty (p.o.), and that (d) the relative expression of FXR only significantly (*/**) increases in the illeum.
- “Fex counters obesity and metabolic syndrome.”
Fex had no effect on chow-fed mice on a normal diet, but on a 60% fat diet (designed to induce obesity), a significant reduction in weight gain was seen, as well as a significant decrease in insuline, cholesterol, leptin and resistin concentrations.
- “Fex improves insuline responsiveness.”
Hyperinsulinemic-euglycemic clamp studies showed that “Fex-treated mice displayed a marked increase in insulin-mediated suppression of HGP compared with control DIO (diet-induced obesity) mice.” Also, “Histological examination of liver tissue from Fex-treated DIO mice revealed a reduction in lipid droplets compared with controls, indicating amelioration of hepatic steatosis.” (“Liver insulin resistance has been linked to obesity-induced hepatic steatosis.”)
- “Fex enhances energy expenditure in brown adipose tissue.”
“Fex-treated DIO mice had consistently higher oxygen consumption (VO₂) and exhaled more carbon dioxide (VCO₂) than vehicle-treated controls.” Increased energy expenditure was also consistent with the observation that “Fex treatment increased the core body temperature of the mice by approximately 1.5 °C.”
Energy expenditure measurements were performed within a Comprehensive Lab Animal Monitoring System (Columbus Instruments) for 6 days, after at least 24h of adaptation time.
It would have been nice if the authors would have also published RER (Respiratory Exchange Rate) values. RER, which is the ratio VCO₂/VO₂, provides information on the active metabolic pathway. RER = 0.7 during the oxydation of carbohydrates and RER = 1.0 during the oxydation of fatty acids. RER ≤ 1.0. During the night, when most humans are asleep, we burn more fat than carbohydrates. For mice, which are nocturnal, the opposite is true.
The higher energy expenditure in brown adipose tissue (BAT) is caused by the greater amount of mitochondria than in white adipose tissue (WAT). Additionally, Ucp1, which is expressed in BAT mitochondria, deflects the energy in the proton gradient to the production of warmth instead of ATP.
- “Fex induces browning of white adipose tissue (WAT).”
The authors report increased Ucp1 expression in adipose tissue. This might (partially) have been induced by increased beta adrenergic receptor (βAR) expression in WAT, through activation by catacholamine, which also explains the increase in FFAs and the decrease in triglycerides.
- “Fex induces fibroblast growth factor 15 (Fgf15) and alters bile acide (BA) composition.”
Fgf15, which is “known to activate the thermogenic program in BAT” (brown adipose tissue), “was found to be markedly upregulated by Fex.”
“Fgf15 also negatively regulates BA synthesis through suppression of hepatic Cyp7a1, which encodes the rate-limiting enzyme for BA synthesis.” “[H]epatic Cyp7a1 was significantly repressed at both the mRNA and protein level after chronic Fex treatment.”
- “Orally active Fex shows minimal systemic exposure.”
-
“What, according to the authors, is the molecular mechanism behind the positive effects of Fexaramine?”
It’s very difficult to say with any certainty at this point how the signalling pathways around Fex-activated FXR are constructed. The authors make no grand claims. They suggest that the increased energy expenditure is caused by Fgf15 activation. But, since systemic FXR agonists, which do robustly induce Fgf15, produce negative side-effects and not nearly as much positive effects, the authors suggest an additional mechanism: the relative increase in the BA composition of lithocholic acid (through the decrease of cholic acid). Lithocholic acid has high affinity for the Tgr5 (=GPBAR1) BA receptor and Gpbar1-/- knockout mice show less marked improvements with Fex treatment.
-
“Do you agree with the conclusions? Is there an alternative interpretation?”
My insight in the affected metabolic pathways is too limited to provide an alternative interpretation.
It is worth noting that, if indeed some of the effects of intestinal-only FXR activation are mediated by lithocholic acid-induced Tgr5 activation, Tgr5 might be a more promising drug target than intestinal FXR. It could be telling of the authors’ FXR/Fex patents that the extra Tgr5 tests have been relegated to the supplementary figures. Hans suspected that these tests might have been done per the request of the reviewers.
-
“What are the weak spots in this research?”
I think that the experimental methods are of themselves sound. I most doubt the objectivity of the researchers concerning the histological comparison of Fex and control tissues, but also their financial interest in the marketability of Fex or another FXR ligand worries me.
Hans emphasized that research should be done on a greater diversity of mouse strains, referring to the caloric restriction (CR) research field [the subject of Tutorial 1], wherein many of the positive results collapsed when testing on some non-inbred mouse strains. A meta-analysis of these studies [citation needed] revealed that CR is more likely to shorten the lifespan of mice than it is to lengthen it. And you have to also take into account that there’s a significant publication bias towards overreporting positive results and agains reporting negative finds, especially if the latter go against spectacular claim made earlier in high-impact journals. The most we can say about CR, for now, is that, depending on the genetic background of a mouse, it may lengthen his or her lifespan.
- “Which other weight-loss medicine do you know? How do these work? Are they effective?”
I know that amphetamines used to be described in the 1950s through 60s. During the lecture on metabolic regulation [Lecture 5], Avandia (rosiglitazone) was mentioned as a drug targetting PPARγ receptors.
- “Which questions remain unanswered in this research? Have new questions been raised by the outcomes? How would you be able to research these?”
During the tutorial, Hans mentioned that what he misses in the article is a study with (intestinal-specific) FXR knockout mice, in which the effects of Fex shouldn’t persist, if indeed the mechanism is as the authors suggest. Cconfronted with this criticism, the authors have responded that FXR knockout-mice are too different, which suggests that maybe they’re not so confident about the intestine-specificity of Fex.
- “Which type of (pre-)clinical follow-up research should/could be done?”
The metabolic pathways involved and the possibility of other drug targets should be investigated first.
- “Why do you think that this article has been published in Nature Medicine?”
The article covers a series of findings, from a number of experiments, giving a very clear direction for further drugs development and testing. If Fex can be less intrusively administered than via oral gavity, it could become an easy, safe weight-loss drugs.
It is remarkable that the article mentions a series of positive physiological/metabolic results. No negative side-effects seem to have been found. Hans mentioned that this might have increased the likelihood of it being accepted by Nature and the popular scientific press. The contrast with the results of systemically acting FXR agonists are suspiciously striking.
- “Finally, how do you rate this article? Give a 1-10 score for the following points:”
“Relevance:” 8/10 “Innovative:” 8/10 “Interesting:” 7/10 “Readability:” 6/10
Update March 30. I incorporated the feedback that we received during our presentation on Friday.

Recent Comments